Improving energy estimation of a chemistry Hamiltonian with SQD¶
In this tutorial we implement a Qiskit pattern showing how to post-process noisy quantum samples to find an approximation to the ground state of a chemistry Hamiltonian: the \(N_2\) molecule at equilibrium in the 6-31G basis set. We will follow a sample-based quantum diagonalization approach to process samples taken from a 36-qubit quantum circuit ansatz (in this case, an LUCJ
circuit). In order to account for the effect of quantum noise, the configuration recovery technique is used.
The pattern can be described in four steps:
Step 1: Map to quantum problem
Generate an ansatz for estimating the ground state
Step 2: Optimize the problem
Transpile the ansatz for the backend
Step 3: Execute experiments
Draw samples from the ansatz using the
Samplerprimitive
Step 4: Post-process results
Self-consistent configuration recovery loop
Post-process the full set of bitstring samples, using prior knowledge of particle number and the average orbital occupancy calculated on the most recent iteration.
Probabilistically create batches of subsamples from recovered bitstrings.
Project and diagonalize the molecular Hamiltonian over each sampled subspace.
Save the minimum ground state energy found across all batches and update the avg orbital occupancy.
For this example, the interacting-electron Hamiltonian takes the generic form:
\(\hat{a}^\dagger_{p\sigma}\)/\(\hat{a}_{p\sigma}\) are the fermionic creation/annihalation operators associated to the \(p\)-th basis set element and the spin \(\sigma\). \(h_{pr}\) and \((pr|qs)\) are the one- and two-body electronic integrals.
The SQD workflow with self-consistent configuration recovery is depicted in the following diagram.
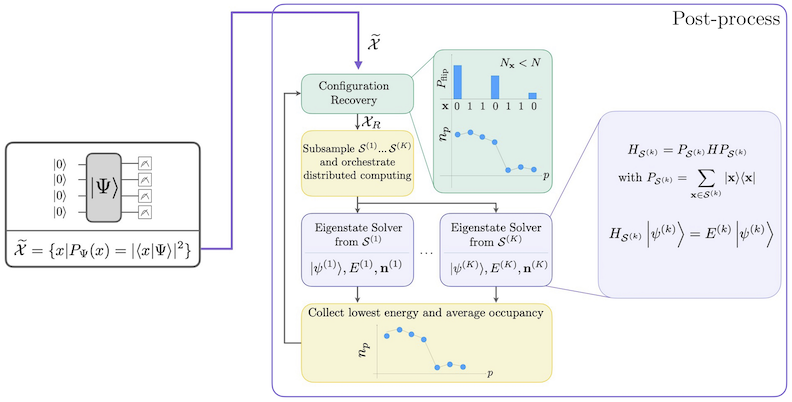
SQD is known to work well when the target eigenstate is sparse: the wave function is supported in a set of basis states \(\mathcal{S} = \{|x\rangle \}\) whose size does not increase exponentially with the size of the problem. In this scenario, the diagonalization of the Hamiltonian projected into the subspace defined by \(\mathcal{S}\):
yields a good approximation to the target eigenstate. The role of the quantum device is to produce samples of the members of \(\mathcal{S}\) only. First, a quantum circuit prepares the state \(|\Psi\rangle\) in the quantum device. The Jordan-Wigner encoding is used. Consequently, members of the computational basis represent Fock states (electronic configurations/determinants). The circuit is sampled in the computational basis, yielding the set of noisy configurations \(\tilde{\mathcal{X}}\). The configurations are represented by bitstrings. The set \(\tilde{\mathcal{X}}\) is then passed into the classical post-processing block, where the self-consistent configuration recovery technique is used. In the SQD framework, the role of the quantum device is to produce a probability distribution.
Step 1: Map problem to a quantum circuit¶
In this tutorial, we will approximate the ground state energy of an \(N_2\) molecule. First, we will specify the molecule and its properties. Next, we will create a local unitary cluster Jastrow (LUCJ) ansatz (quantum circuit) to generate samples from a quantum computer for ground state energy estimation.
First, we will specify the molecule and its properties.
[1]:
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Next, we will create the ansatz. The LUCJ ansatz is a parameterized quantum circuit, and we will initialize it with t2 and t1 amplitudes obtained from a CCSD calculation.
[2]:
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
We will use the ffsim package to create and initialize the ansatz with t2 and t1 amplitudes computed above. Since our molecule has a closed-shell Hartree-Fock state, we will use the spin-balanced variant of the UCJ ansatz, UCJOpSpinBalanced.
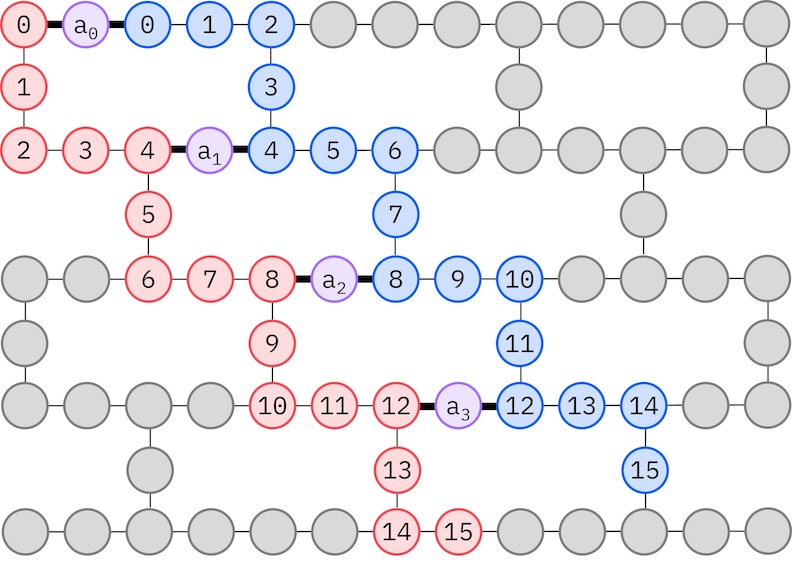
As our target IBM hardware has a heavy-hex topology, we will adopt the zig-zag pattern for qubit interactions. In this pattern, orbitals (represented by qubits) with the same spin are connected with a line topology (red and blue circles) where each line take a zig-zag shape due the heavy-hex connectivity of the target hardware. Again, due to the heavy-hex topology, orbitals for different spins have connections between every 4th orbital (0, 4, 8, etc.) (purple circles).
[3]:
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Step 2: Optimize the problem¶
Next, we will optimize our circuit for a target hardware. We need to choose the hardware device to use before optimizing our circuit. We will use a fake 127-qubit backend from qiskit_ibm_runtime to emulate a real device. To run on a real QPU, just replace the fake backend with a real backend. Check out the Qiskit IBM Runtime docs for more info.
[4]:
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Next, we recommend the following steps to optimize the ansatz and make it hardware-compatible.
Select physical qubits (
initial_layout) from the target hardware that adheres to the zig-zag pattern described above. Laying out qubits in this pattern leads to an efficient hardware-compatible circuit with fewer gates.Generate a staged pass manager using the generate_preset_pass_manager function from Qiskit with your choice of
backendandinitial_layout.Set the
pre_initstage of your staged pass manager toffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITincludes Qiskit transpiler passes that decompose gates into orbital rotations and then merges the orbital rotations, resulting in fewer gates in the final circuit.Run the pass manager on your circuit.
[5]:
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Step 3: Execute experiments¶
After optimizing the circuit for hardware execution, we are ready to run it on the target hardware and collect samples for ground state energy estimation. As we only have one circuit, we will use Qiskit Runtime’s Job execution mode and execute our circuit.
Note: We have commented out the code for running the circuit on a QPU and left it for the user’s reference. Instead of running on real hardware in this guide, we will just generate random samples drawn from the uniform distribution.
[6]:
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Step 4: Post-process results¶
Now, we run the SQD algorithm using the diagonalize_fermionic_hamiltonian function. See the API documentation for explanations of the arguments to this function.
The solver included in the SQD addon uses PySCF’s implementation of selected CI, specifically pyscf.fci.selected_ci.kernel_fixed_space. The example below also shows how to pass keyword arguments to that function via the included solver. Here we pass the max_cycle argument.
[7]:
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
Now, we plot the results.
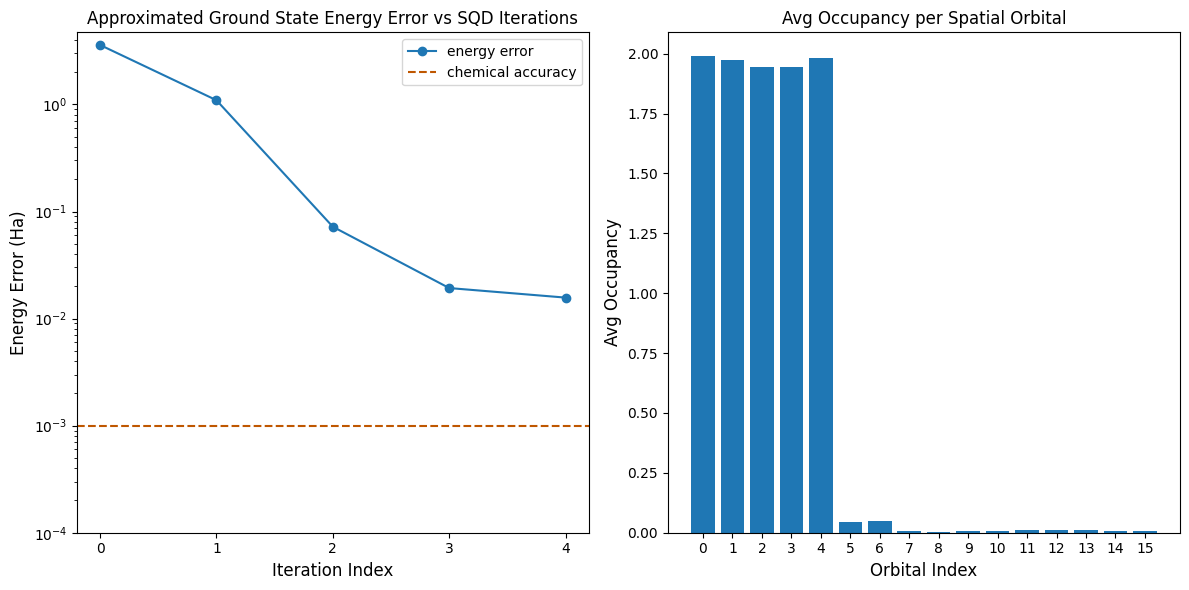
The first plot shows that after a few iterations we estimate the ground state energy within ~16 mH (chemical accuracy is typically accepted to be 1 kcal/mol \(\approx\) 1.6 mH). Remember, the quantum samples in this demo were pure noise. The signal here comes from a priori knowledge of the electronic structure and molecular Hamiltonian.
The second plot shows the average occupancy of each spatial orbital after the final iteration. We can see that both the spin-up and spin-down electrons occupy the first five orbitals with high probability in our solutions.
[8]:
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha